Molecular Modeling
Atomic structure and dynamics of melts and fluids
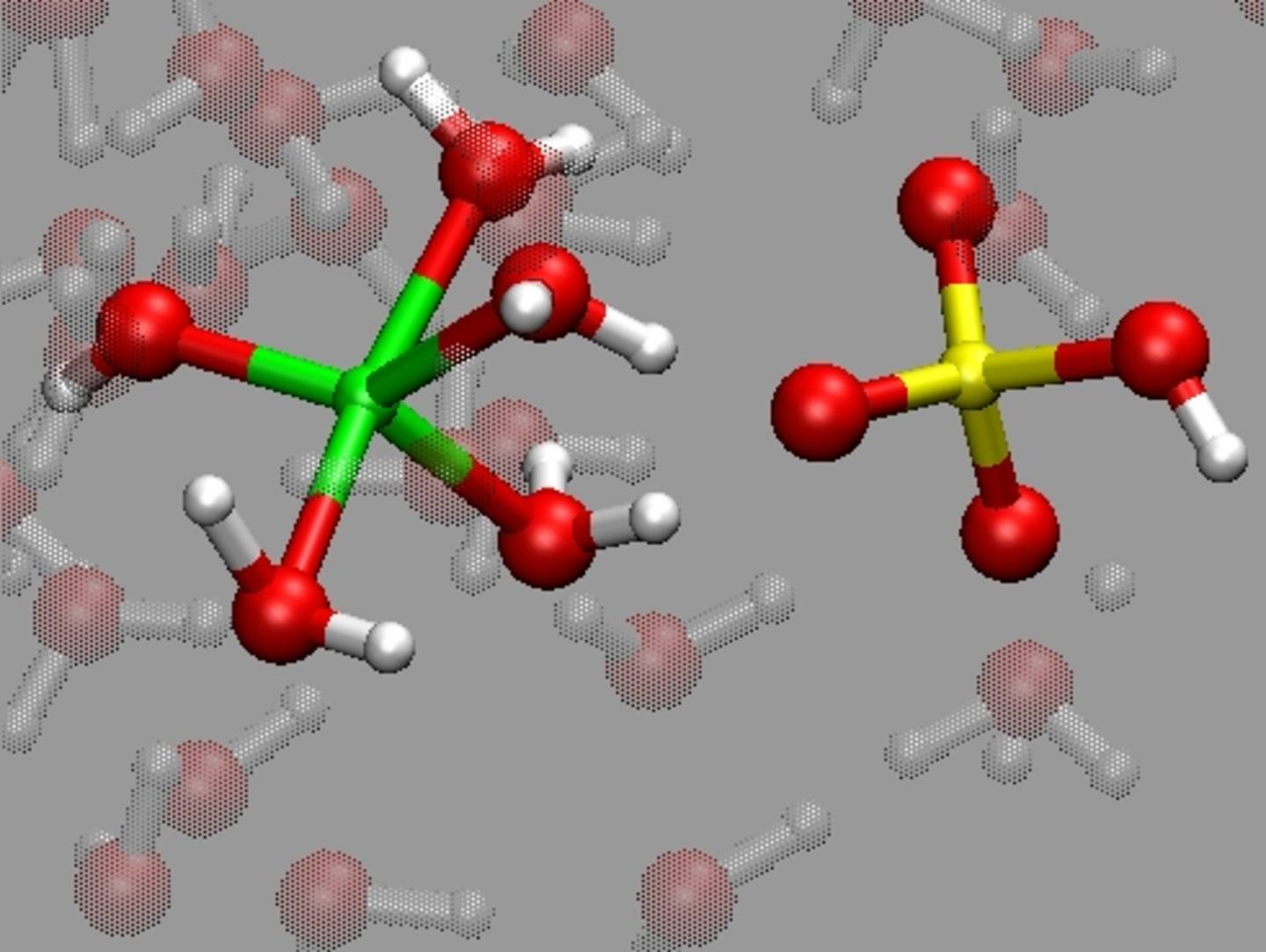
Melts and fluids are involved in most geological processes in the Earth's crust and mantle. We are interested in how the atomic structure and dynamics influence their physical properties, such as density, viscosity, electrical conductivity or sound velocity. We also study the chemical speciation and hydration in aqueous fluids, which are important parameters for thermodynamic modeling.
We perform molecular dynamics (MD) simulations using both classical and quantum mechanical models for the particle interactions. The latter so called ab initio molecular dynamics simulations are performed on supercomputers.
Realistic models are not only needed to interpret experimental data but also to predict properties of melts and fluids at extreme conditions of pressure and temperature that are not accessible experimentally. The construction of such models is a big challenge in (geo)materials research.
Mineral properties at high pressure and/or high temperature
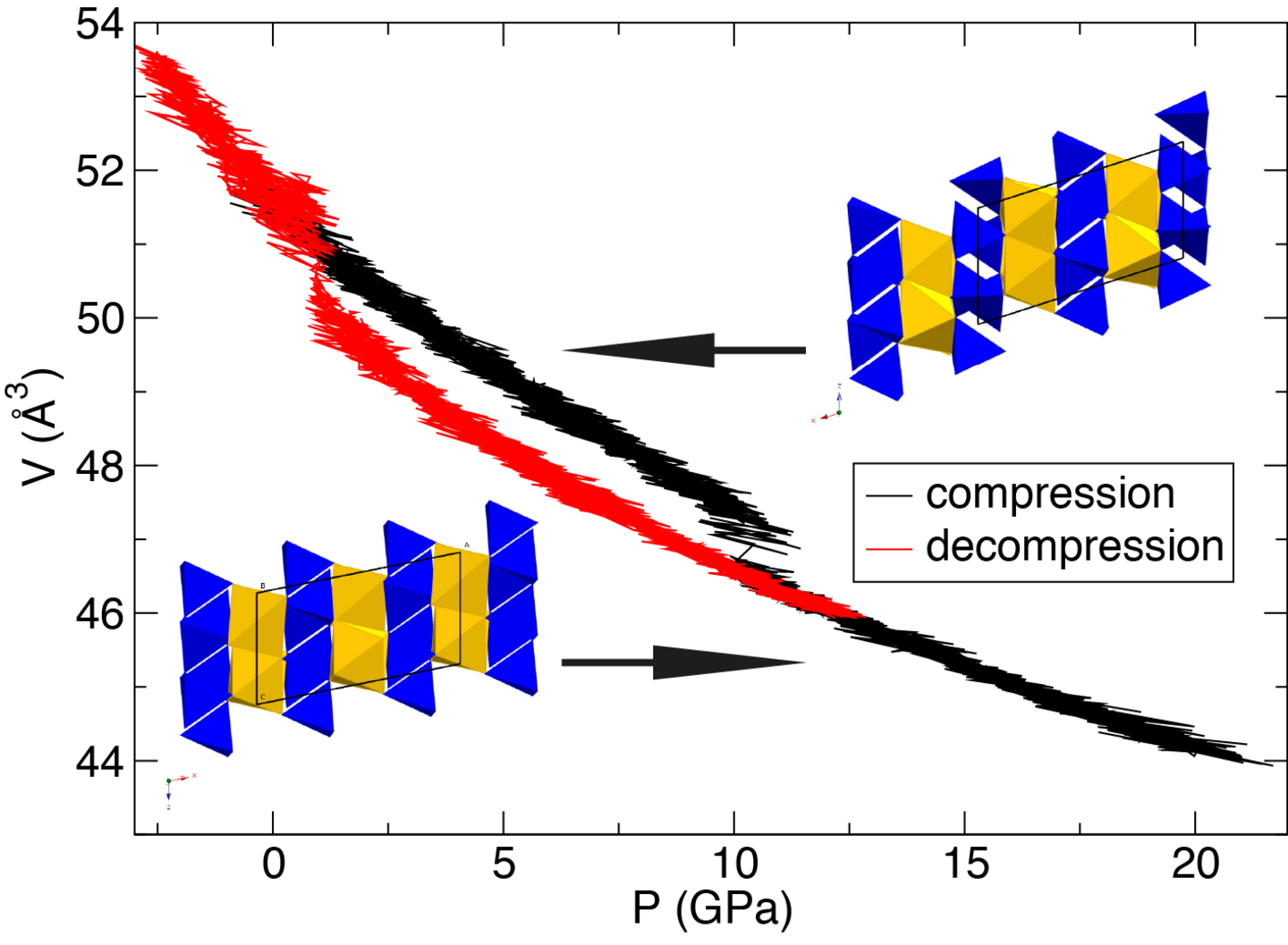
We use first-principles methods based on density functional theory (DFT) to study the structural, physical and thermodynamic properties of minerals. For instance, static energy calculations allow us to compare the relative stability of polymorphic phases at different thermodynamic conditions. Using molecular dynamics (MD) and metadynamics simulation techniques, we study the mechanisms and the kinetics of mineral phase transformations, diffusion processes in minerals or thermoelastic properties of minerals.
Structure and transport at grain and phase boundaries

Properties of solids and liquids close to interfaces are often different from the bulk. Also, most geochemical processes such as chemical reactions or material transport are somehow related to interfaces. We are involved in a number of projects looking e.g. at the atomic structure of mineral grain boundaries, melting and crystallization processes, or material transport along grain boundaries.
Atomistic modeling tools
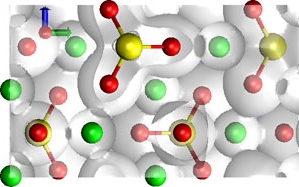
The biggest challenge for atomistic modeling is the development of accurate and efficient ways to represent particle interactions. With the fast growing availability of computing power, electronic structure calculations based on density functional theory (DFT) have become the most powerful tool for applications in geomaterials modeling. We are using DFT codes (e.g. CPMD, ABINIT) to study static and dynamic properties of minerals, melts and fluids.
For large scale simulations (number of atoms >> 1000 and timescales >> 100 ps) as needed e.g. to study properties of melts, defect crystals or interfaces we still need to use classical interaction potentials. Advanced ionic interaction models such as the Aspherical Ion Model (AIM) that is parameterized using first-principles calculations are capable to describe accurately the properties of silicates in a wide range of compositions, pressures and temperatures as required for geomaterials modeling. We are actively participating in the improvement of such models.