Molekulare Modellierung
Atomare Struktur und Dynamik von Schmelzen und Fluiden
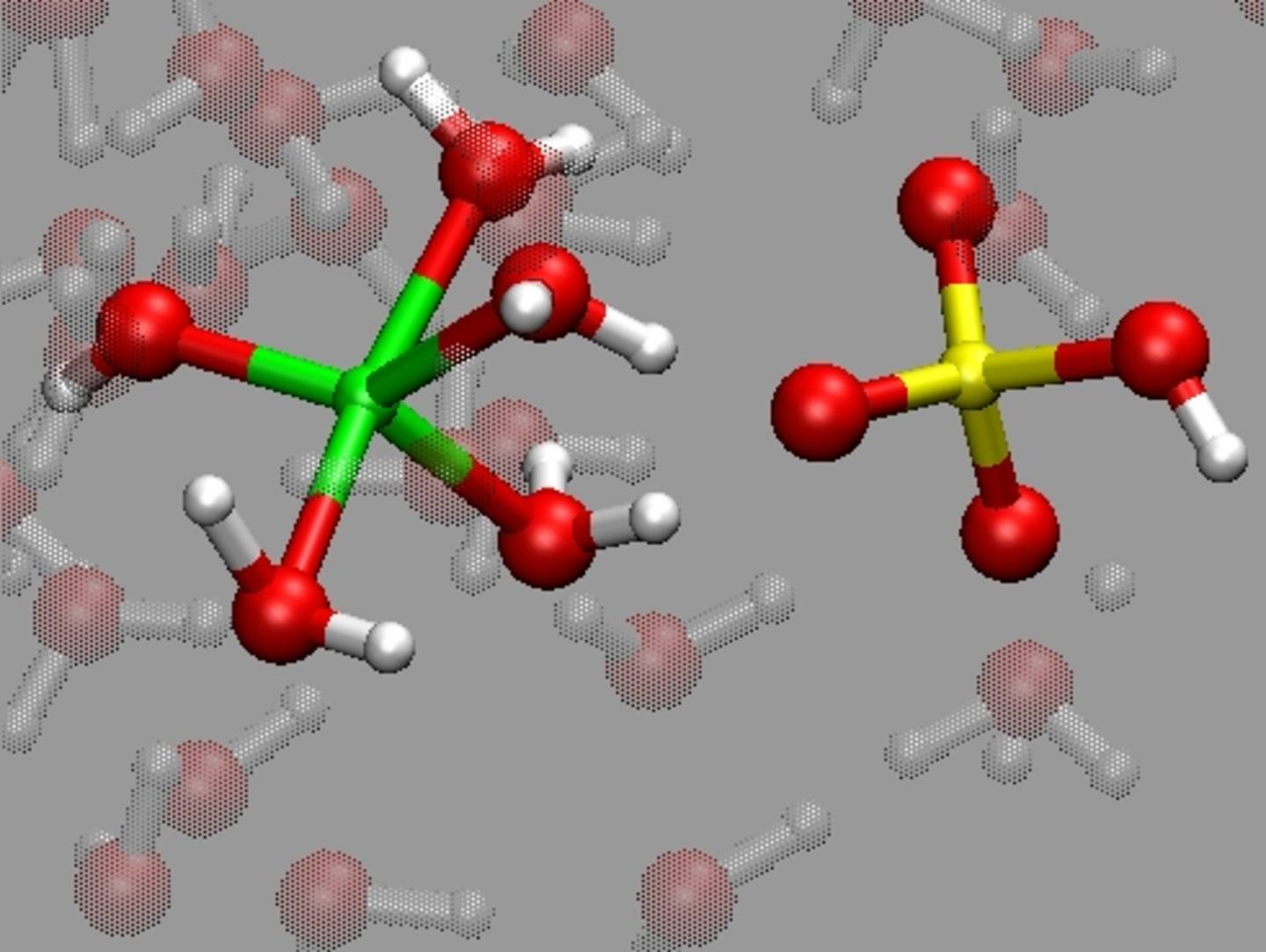
Schmelzen und Fluide spielen eine wichtige Rolle in den meisten geologischen Prozessen in der Erdkruste und im Erdmantel. Wir untersuchen, wie die atomare Struktur und Dynamik von Schmelzen und Fluiden die entsprechenden physikalischen Eigenschaften (z.B. Dichte, Viskosität, elektrische Leitfähigkeit oder Schallgeschwindigkeit) beeinflussen. Wir interessieren uns außerdem für die chemische Speziation und Hydratation in wässrigen Fluiden, die wichtige Parameter für die thermodynamische Modellierung darstellen.
Methodisch nutzen wir sowohl klassische als auch ab initio Molekulardynamik (MD) Simulationen. Da letztere sehr rechenaufwändig sind, werden diese Simulationen auf Supercomputern durchgeführt.
Realistische Modelle werden nicht nur zur Interpretation experimenteller Daten benötigt, sondern auch zur Vorhersage von Schmelz- und Fluideigenschaften bei extremen Druck- und Temperaturbedingungen, die experimentell nicht zugänglich sind. Die Konstruktion solcher Modelle stellt eine große Herausforderung in der (Geo-) Materialforschung dar.
Mineraleigenschaften bei hohen Drücken und/oder hohen Temperaturen
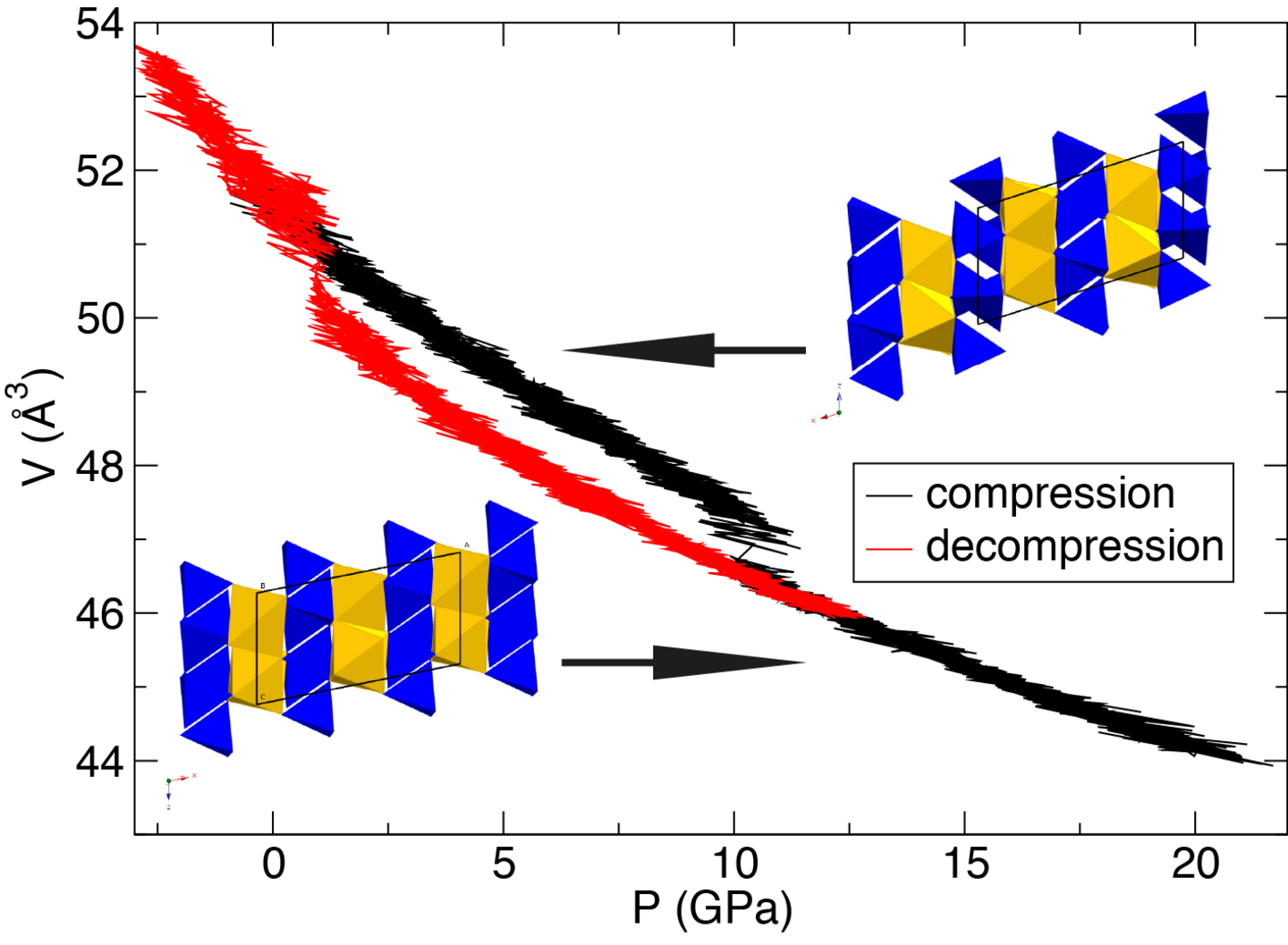
Wir verwenden ab-initio Methoden zur Untersuchung struktureller, physikalischer und thermodynamischer Mineraleigenschaften. Zum Beispiel können aus statischen Energieberechnungen die relativen Stabilitäten polymorpher Phasen bei unterschiedlichen thermodynamischen Bedingungen verglichen werden.
Um die Mechanismen und die Kinetik von Phasentransformationen atomistisch zu verstehen, werden Methoden der Molekulardynamik (MD) und der Metadynamik kombiniert. In MD-Simulationen untersuchen wir außerdem Diffusionsprozesse in Mineralen oder thermoelastische Mineraleigenschaften.
Struktur und Transport an Korn- und Phasengrenzen von Mineralen

An Grenzflächen unterscheiden sich die Eigenschaften von Festkörpern oder Flüssigkeiten of stark von denen im Volumen. Dabei finden die meisten geologischen Prozesse, wie z.B. chemische Reaktionen oder Materialtransport an Grenzflächen statt. Unsere Forschungsaktivitäten in diesem Bereich konzentrieren sich auf die atomare Struktur von Mineralkorngrenzen und anderen Kristalldefekten, auf Schmelz- und Kristallisationsprozesse sowie auf Materialtransport entlang der Korngrenzen.
Methodenentwicklung, besonders im Bereich klassischer Wechselwirkungspotenziale
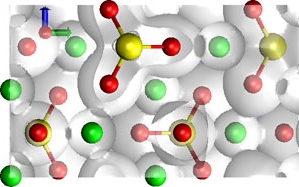
Eine der größten Herausforderungen für die atomistische Modellierung ist die Entwicklung akkurater und effizienter Methoden zur Beschreibung der interatomaren Wechselwirkung. Durch den rasanten Zuwachs an Rechenkapazitäten haben sich elektronische Strukturberechnungen auf Grundlage der Dichtefunktionaltheorie (DFT) in den vergangenen Jahren zur leistungsstärksten Methode im Bereich Geomaterialmodellierung entwickelt. Wir nutzen Standard-DFT Codes (z.B. CPMD, ABINIT) zur Untersuchung statischer und dynamischer Eigenschaften von Mineralen, Schmelzen und Fluiden.
Die Modellierung von Schmelzen, Defektkristallen oder Grenzflächenprozessen erfordert große Simulationszellen (Anzahl der Atome >> 1000) und lange Simulationszeiten (>> 100 ps), sodass dafür klassische Wechselwirkungspotenziale eingesetzt werden müssen. Komplexe ionische Wechselwirkungsmodelle, wie das Aspherical Ion Model (AIM), das mit Hilfe von ab initio Methoden parametrisiert wird, sind in der Lage, die Eigenschaften von Silikaten in einem weiten Druck-, Temperatur- sowie chemischen Zusammensetzungsbereich akkurat zu beschreiben. Wir sind aktiv an der Verbesserung solcher Modelle beteiligt.